{kind=link}
Science & Nature
From made-to-order genetic therapies to model organisms engineered to be ‘patient avatars’, the technology exists right now to save patients with rare diseases.

PM Images / DigitalVision / Getty
Last October, Susannah Rosen, an 8-year-old girl with a devastating neurodegenerative genetic disease, took her first dose of a custom-made genetic medicine. What’s more, she got it at no cost, provided by the non-profit foundation n-Lorem. The drug isn’t gene therapy: it doesn’t change Susannah’s genome or undo her mutation. It’s an antisense oligonucleotide (ASO), a short sequence of modified DNA, designed to bind to a problematic mRNA transcript and stop production of the disease-causing protein.
Susannah joins a small but growing group of patients who have received treatments designed just for them, called ‘N of 1’ therapies. Personalized medicine seemed to be just around the corner for decades, but in 2018, the corner had finally been turned when a custom-designed ASO made its clinical debut at Boston Children’s Hospital. Researchers there, led by Tim Yu, developed an ASO drug called milasen to treat a child — Mila — who had a one-of-a-kind genetic disorder. Incredibly, that drug was designed, manufactured and administered in less than a year1.
ASOs are only one approach to designing N-of-1 drugs; CRISPR gene editing, an inherently customizable tool, is moving successfully into the clinic, making it another promising candidate. Clinical trials underway for genetic diseases including sickle cell anemia and congenital blindness, though these are not ultra-rare conditions, nonetheless have demonstrated that the technique can be used safely in humans. Some people are pushing to apply personalized CRISPR therapies more broadly, but regulators remain cautious. Once gene editing is performed, it can’t be reversed if adverse events arise.
Another way that CRISPR can contribute is by creating models for drug repurposing studies. By genetically modifying yeast or other model organisms to mimic a patient’s mutation, researchers can screen existing drugs to see whether any address the defect causing a single patient’s disease.
What all these strategies have in common is the issue of cost. Without a large customer base to buy the resulting treatments, scientists working on rare diseases are struggling to build new models that enable access to life-saving treatments for all who need them.
Balancing safety with speed
ASOs aren’t new; the US Food and Drug Administration (FDA) approved the first, fomivirsen, in 1998. Developed as an antiviral against cytomegalovirus retinitis in people with AIDS, fomivirsen provided a proof-of-concept for clinical ASOs. It would be 15 years before antisense drugs began to take off. Since 2013, however, no fewer than eight ASO drugs have been green-lighted for use in the United States or Europe to treat genetic diseases2, including Spinraza (nusinersen), the first FDA-approved therapy for spinal muscular atrophy (SMA), a motor neuron disease that, in its most severe form, causes death by age two.
Spinraza’s development, like that of so many therapeutics, was a multi-year odyssey, beginning in 2004 and not ending until the drug won approval in 2016. In contrast, the milasen story showed that the timeline can be accelerated. Because Mila’s disease was progressive and fatal, the risks of waiting a year for animal safety studies outweighed the danger of possible adverse effects from the treatment, so Mila began treatment while the animal safety study was still in progress. Her treatment was authorized by the FDA under an expanded-access protocol, which allows a patient with a life-threatening condition to be treated with an investigational drug outside of clinical trials. However, exactly where to draw the line between letting a child suffer from a degenerative disease and trying a potentially harmful experimental treatment remains a subject of vigorous debate. In a 2019 editorial in the New England Journal of Medicine, the same issue where the development of milasen was published, Janet Woodcock and Peter Marks at the FDA raised issues about how to proceed safely, arguing that “even in rapidly progressing, fatal illnesses, precipitating severe complications or death is not acceptable”3.
Since then, the FDA has published a non-binding guidance document for investigators developing personalized ASO drugs. It lays out protocols for dose escalation schedules, regular safety assessments, and the establishment of stopping criteria in case of serious adverse events. The document explicitly does not address the level of preclinical data or drug quality requirements needed to start treating a patient, and it also excludes drugs intended for commercial marketing.
“You can make a really strongly ethical case that if something is safe in rats, and a patient is going to die, or is going to lose neurons, it’s a better move for that patient to proceed than to wait for a monkey study, because the risk of not treating outweighs the risk of treating,” says Jonathan Watts of the RNA Therapeutics Institute at the University of Massachusetts Medical School.
Yu points out that the ethical issues around personalized ASOs aren’t unique. “Through these guidances, we are seeing concepts that are already in clinical practice and regulatory practice in fields like oncology, where patients who have failed first, second, third, fourth line therapy are often first subjects in very early stage, investigational trials of quite new agents,” Yu says. “Patients and their clinicians weigh their decisions to participate in these trials knowing their disease is fatal and other options have been exhausted.”
Regulatory policies differ between the United States and Europe, points out Annemieke Aartsma-Rus, a translational geneticist at Leiden University Medical Center in the Netherlands, and this affects what types of mutations are eligible for N-of-1 designation. “There’s two things you can do with ASOs: restore a protein or reduce the production of a toxic protein,” she says. An ASO that treats a disease by reducing a toxic protein could conceivably work for anyone who makes it, regardless of the specific mutation. “You can develop it for one individual, but you know it applies to others,” explains Aartsma-Rus. “In the US, you are allowed to say ‘we’re going to develop this in an N-of-1 setting’, but in Europe, they would say no, it’s not allowed. You either do it for everyone who’s eligible, or not at all — not for one specific patient who happens to be able to fund that.”
Milasen is an exon-skipping ASO. It is designed to bind to a mis-spliced section of the mRNA for MSFD8 that interferes with the production of a lysosomal transporter protein needed for neuronal survival in an ultra-rare form of Batten disease (a late infantile neuronal ceroid lipofuscinosis, CLN7)4. “A cryptic splicing mutation leads to the inclusion of part of an intron in the messenger RNA, and that disrupts protein production,” Aartsma-Rus explains. “If you can skip that cryptic exon, the normal transcript is back, you can restore the normal protein.” A properly designed ASO can do just that: neutralize a cryptic exon, restoring the normal transcript. Thus, she says, these types of mutations are the most promising avenue for N-of-1 therapies because cryptic splicing mutations are unique and the tailor-made ASO will work in only one patient. However, mutations occurring in introns are difficult to identify because introns contain a lot of sequence variability. It’s not always obvious whether a particular variant is disrupting the transcript. “People say [cryptic splicing mutations] are very rare, but the reality is they’re probably not that rare, it’s just really difficult to find them,” says Aartsma-Rus.
Aartsma-Rus serves as chair of the Dutch Center for RNA Therapeutics, a nonprofit consortium of medical centers developing personalized RNA therapies. Identifying an appropriate mutation is just the first issue, she says (Fig. 1). Ideally, one would want to apply it to a disease that affects the brain or the eye — someplace the ASO can be injected locally. “You can use a very low amount, and you don’t have to worry about systemic exposure,” she says. She adds that there must be some hope of benefit to the patient at the time of treatment. “You can do local injection in the eye, but if someone is totally blind, you can restore the protein, but they will stay blind,” she says. “You’re not going to return function into the cells. If the disease is too advanced, the patient won’t benefit.” The ultimate decision rests with the patient, she emphasizes. It’s critically important that the patient and the family understand realistically what benefits they can expect and the treatment risks.
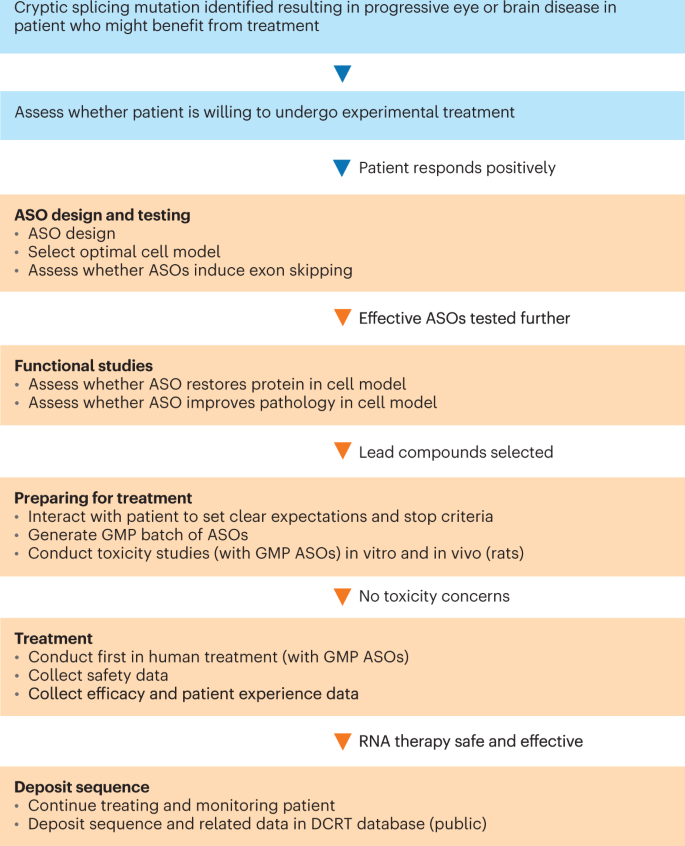
ASOs are under development for patients with an ultra-rare (fewer than 100 worldwide) serious, life-threatening diseases who have no therapy available, and where there is no commercial incentive to develop therapies. Source: Dutch Center for RNA Therapeutics (DCRT). ASO, antisense oligonucleotide; GMP, good manufacturing practices; IND, Investigational New Drug.
N of 1 … and another, and another
Unlike in Europe, in the United States “N of 1” can mean “N of 1 at a time.” Jacifusen is an ASO designed against the FUS gene (RNA binding protein fused in sarcoma), which, when mutated, causes an aggressive form of amyotrophic lateral sclerosis (ALS). It’s named for Jaci Hermstad, who in 2019 became the first patient to receive the drug, but it wasn’t designed specifically for her. The drug was already being developed by Ionis and collaborators at Columbia University and had shown efficacy in a Fus-mutant mouse model5 when Hermstad received her diagnosis at age 25. Her twin sister, Alex, had died of the disease when she was 17, and, terrified of losing their remaining daughter, Jaci’s parents launched a campaign to get permission from the FDA for Jaci to try the drug, even though it hadn’t yet been tested in humans. At the time Jaci’s disease was diagnosed, the FDA had not yet released their draft guidance around the use of individualized ASOs in patients.
“The FDA were extremely responsive, thoughtful, reasonable partners in this,” says Neil Shneider of Columbia, who developed jacifusen. “Their goal was clearly to protect my patient but at the same time recognize the desperate nature of the disease and the risks the disease presented to her.” Although Shneider had limited toxicity data on jacifusen, the drug was chemically similar to two other Ionis ASOs for ALS that had already been introduced into the clinic. Jaci started treatment under an expanded-access Investigational New Drug (IND) application, and after that, more patients with FUS-related ALS wanted to try it. “The program grew, but it was incremental, one by one,” Shneider says. “We ultimately submitted 12 INDs in total for the same therapeutic.” After the first few patients, the FDA set limits on the serial IND approach, urging Shneider to conduct a clinical trial. “The program might have ended, but it was saved by Ionis,” he says. “They agreed to sponsor a proper clinical trial and they did it very quickly.”
Shneider expects to publish the data on the 12 IND patients in 2023; although Jaci passed away about a year after starting the drug, “she showed encouraging signs that she was responding to the ASO,” Shneider says
Data sharing
After milasen, Yu began hearing from families hoping that antisense drugs could help their children. Mindful of the ethical considerations involved, Yu decided to take on a devastating genetic disease, ataxia–telangiectasia, creating a drug called atipeksen for a 3-year-old patient named Ipek in October 2019. Ipek is doing well 3 years into treatment, Yu says, but the results have not yet been published. In 2020, he treated two toddlers with a severe form of epilepsy with an ASO called valeriasen. Although the drug reduced the frequency of the seizures in one patient and eliminated them in the other, both children developed hydrocephalus and subsequently stopped treatment. One eventually died (of causes unrelated to the treatment); the other has seen her seizures increase in frequency since stopping treatment, and her parents are considering restarting it.
The unexpected adverse event underscores the importance of data sharing, which Yu emphasizes as key to advancing the field. “The moment we encountered hydrocephalus in our patients, we contacted everybody in the field we knew that worked with ASO drugs,” Yu says. “People have been studying ASOs, including extensive animal testing, for 30 years. Everyone said, ‘No, we’ve never seen this.’ So it’s something that we have to work on.” Although it hadn’t been seen in animal studies, however, hydrocephalus had occurred previously in people treated with an ASO. In March 2021, a phase 3 trial for the drug tominersen, developed by Ionis for patients with Huntington’s disease, was prematurely halted due to “ventricular expansion” — essentially, hydrocephalus — which was observed in a dose-dependent fashion. Testing of tominersen has resumed under the auspices of Roche, which is partnering with Ionis.
Yu says the most likely hypothesis is that the fluid buildup is a class effect, but the mechanism responsible for it remains unclear. “It would be really nice if we could simply just get some tominersen and get some valeriasen and do the appropriate experiments to try to identify mechanistic commonalities between the two sequences,” he says. “That isn’t exactly how the world always works. But the sooner we can get to a point where we’re doing those sorts of things in a collaborative way, the better for everybody.”
Together with Julia Vitarello, the mother of Mila, Yu launched the N=1 Collaborative, or N1C, an organization dedicated to connecting ASO investigators around the world and providing a hub for communication and data sharing. Yu points out that, often, barriers to data sharing arise as unintended consequences of the way medical data are handled and patients’ privacy is protected. “One of the critical things that I’m hoping the N=1 Collaborative can do is nip some of that in the bud,” Yu says, “and set sharing standards that make it easy for investigators and the families they work with to volunteer for their data to be shared and aggregated in a way that informs the field.”

Tim Yu of Boston Children’s Hospital is helping to create a centralized resource to help streamline ASO development for N-of-1 diseases.
Credit: Michael Goderre, Boston Children’s Hospital
“The platform technologies are continuing to improve, and I would say right now, the therapeutic index is reasonably narrow,” says Watts. “We can find sequences that are well tolerated and that save lives. But there are also definitely examples that are quite toxic of these ASOs of different sequences with the same chemistry. There are examples that are a little bit borderline — they have some beneficial effect, but it’s not clear whether they’re safe enough yet. I think if we could get the platform to have a therapeutic index that was ten times greater, it would be so much easier to do single-patient drugs.” The way to get there, Watts says, is through collaboration and sharing of data.
N1C also sponsors workshops to help the field develop consensus on certain best practices, including guidelines for mutation selection, preclinical testing, safety tests, regulatory issues, and metrics to define how well a treatment is working. The organization also serves as a resource for doctors who might not have experience in genetic therapies. Last year the group published consensus guidelines6 for designing and testing ASOs, and they are putting together an online database of control ASOs confirmed to work well in various cell types.
“A lot of clinicians might have a patient with a rare disease and want to help, but don’t have any idea how to approach the FDA. Where do you find a source of oligonucleotide for under $500,000? For a single patient?” Watts says. “Sharing information and help, guidance and resources and protocols around all of those kinds of questions is another thing that the more clinically minded people in the collaborative do for each other.”
One major player in ASOs has so far declined to share data in the collaborative: n-Lorem.
Giving away ASOs for free
Stanley Crooke, founder of Ionis, recognizing that the traditional for-profit pharma company model couldn’t support developing N-of-1 ASOs, founded the n-Lorem Foundation in 2020. n-Lorem’s mission is to provide experimental ASO treatments to people with genetic diseases that affect fewer than 30 patients worldwide. The foundation was launched with personal funding from Crooke and Rosanne Crooke, his wife, plus $3.4 million from Ionis and $1.75 million from Biogen. Crooke says that other companies are stepping up to contribute as well. “We’ve had wonderful response,” Crooke says. “Really every vendor in the oligonucleotide industry is giving either free or deeply discounted work, and often cash donations as well.” They’ve also received donations from disease advocacy organizations.
Crooke also has kind words for the FDA in terms of how they’ve handled the personalized therapies. “Being fast is important. Being high quality is even more important. And it’s the combination of the efficiency and versatility of antisense technology with the special guidance that the FDA has issued for the treatment of nano-rare patients with ASOs that allow us to move as fast as we are,” he says.
At the same time, he resists the idea that n-Lorem should contribute their data to the N1C efforts. Pointing out that Ionis has been “perfecting” ASO technology since 1989, he says it wouldn’t make sense to compare their data with that of others just starting out. “We offer to collaborate with basically every investigator who approaches us,” he says. “Our collaborations are that we design and make the ASOs, and then we bring an optimal ASO forward, and then the investigator treats the patient.”
Crooke expressed support for the idea of establishing databases, as long as minimum standards for preclinical testing processes and performance are defined and rigorous clinical evaluation protocols with well-defined clinical endpoints are agreed on. “As such, n-Lorem is open to contributing data to the N=1 Collaborative or other data platforms as appropriate,” he says. But, because N1C pools data from different investigators all using different methods to make ASOs, Crooke says, “one of the worries that I have is that this is being taken on by people who don’t really understand this technology.”
Mining for hidden gems
ASOs are not the only route to N-of-1 therapies. CRISPR can help people with ultra-rare diseases. Rather than changing a patient’s DNA, the technology enables researchers to create yeast or worm models with genetic errors that match those in the patients, thus creating a personalized resource for high-throughput screening of small-molecule drug candidates. “You don’t program the medicine, like in the ASO model; you program the yeast model,” explains Ethan Perlstein, CEO and founder of Perlara, a public benefit corporation (Box 1). “What has a faster path to the clinic than even a customized ASO? A repurposed drug that already has a safety record.”
Perlstein says the idea behind Perlara is to “productize” drug repurposing by using existing yeast, worm and fly models. The company is actually on its second iteration, having been founded originally in 2014 before shutting its doors in 2019 because of lack of funding. Yet some important unfinished business lingered. For two years, Perlara had been working with Maggie’s Cure, an LLC formed by the family of Maggie Carmichael. Maggie has a congenital disorder of glycosylation (CDG), PMM2-CDG, that affects multiple organ systems. Researchers working with Perlara were already screening yeast and worm models of Maggie’s disease, looking for a drug that might be effective. The project continued after the company officially closed, and in January 2020, Maggie began a drug called epalrestat under the supervision of doctors at the Mayo Clinic in Rochester, Minnesota. The drug is not FDA-approved for any indication, but it is approved in Japan as a treatment for diabetic neuropathy. Since she began taking epalrestat, Maggie’s motor skills have improved and her vocabulary has grown dramatically.
Around the time that Maggie was starting on epalrestat, another family was searching for answers for their son’s CDG. Jake Carroll was diagnosed with Man1b1-CDG in early 2020, and his parents, Claire Fast and Matt Carroll, soon discovered how little was known about the disease. “It was quite a helpless feeling,” Fast says. “Very few people in the world know about this disorder.” The Carrolls met the Carmichaels in a Facebook group for families affected by CDG, and that’s when they learned about Perlara. In 2022, Perlstein connected the family with Clement Chow at the University of Utah. Chow works on CDGs in fruit flies, and he agreed to conduct a screen for Man1b1 drugs in flies.

Six-year-old Jake Carroll has an ultra-rare metabolic disorder called Man1b1-CDG. Perlara helped his parents find a researcher who could make a fruit fly model of his disease for drug screening.
Credit: Carroll family
“We were so desperate at that point,” Fast recalls. “Learning about what Perlara was doing was very exciting for us, just to find somebody who knew how to navigate the research. It’s really hard to know where to start.”
Chow’s fly models did not carry an exact replica of Jake’s mutation, but rather a general Man1b1 knockdown made using RNA interference (RNAi). “In the future, we may want to move toward making more personalized models, using CRISPR to put in real patient mutations, but that takes so much more time,” he says. “We can order up all the reagents to do this RNAi knockdown experiment right after the call with the patients, get the reagents within a week, have a few tests done in about three weeks, and then it’s off to the races. If we do CRISPR, it’s months before we even have a fly we can test.”
As of January 2023, the screen has identified several drugs, and the next step will be to sit down as a team and discuss how to proceed. “About 20% of the hits we found in our screen all fell into the same category, so that was really encouraging,” Chow says. “A number of them either have a pretty good history in pediatric administration or they’re actually OTC [over the counter].” Once a drug is selected, theoretically, Jake could begin treatment right away. Indeed, another Perlara patient named Lucy, who suffers from a different CDG, PGAP3, received her first dose 48 hours after her yeast screen was completed. In that case, the family was working with two labs in parallel: while one performed the yeast screen, the other was studying Lucy’s own cultured cells, and both teams identified the same biochemical pathway. Within a week of starting her drug regimen, Lucy took her first unassisted step.
The vast collection of small-molecule drugs that have already been safety-tested represents an underutilized resource that can be mined for cures. “We’re banking on being able to find all these hidden gems amongst the things we’ve already approved as safe,” says Chow. “I think that it will work for a large number of diseases. There’s just so much biology hidden in these drugs. Most of them, we don’t really have a good sense of the exact mechanism of action.”
Perlstein says he has around 30 clients looking for treatments. “Drug repurposing is going to go exponential soon,” Perlstein predicts. “What were these few N-of-1s is going to become a tide of N-of-1s. And, of course, that’s what they talk about with ASOs, that’s what n-Lorem is meant to accommodate, and the N=1 Collaborative. They’re trying to get ahead of this tsunami. And it’s coming.”
Can rare disease drugs be profitable?
One year ago, the US National Institutes of Health (NIH) launched its Ultra-rare Gene-based Therapy (URGenT) program to accelerate the clinical development of therapies for neurological diseases7. “It’s still under construction,” says program director Chris Boshoff. “At the moment, it only consists of a preclinical component. There are two funding opportunity announcements for projects up to and including an IND filing.” A clinical network is in development and projected to be up and running sometime in 2023, he says. “That will really change the dynamics of this program. We hope it will provide almost a seamless transition from preclinical to clinical, and a streamlined access process. If applicants already have an IND, they don’t have to go through the same lengthy application process for funding.” The clinical network database will also serve as a data sharing resource, which can help other researchers. “Even if it’s as small as just sharing a clinical trial protocol, or sharing assays during development, it’s a start,” Boshoff says.
No projects have been funded so far, but Boshoff says the scope of the program encompasses any gene-based or transcript-directed technology, including ASOs, gene editing, or classical AAV-based gene therapy, provided they address ultra-rare neurological diseases.
In a recent New York Times editorial, Fyodor Urnov, scientific director at the Innovative Genomics Institute and professor at the University of California Berkeley, suggested that for rare diseases, public funding makes more sense than for-profit businesses, pointing to the financial struggles of two flagship gene therapy companies, bluebird bio and Editas Medicine, as evidence8. By contrast, he says, “the California Institute for Regenerative Medicine has consistently and vigorously supported a path to the clinic for experimental therapies including CRISPR” and is the only such state-funded program to do so. At the federal level, he says, the NIH offers funding for academic researchers to develop CRISPR-based medicines. “I think this is exactly the right approach,” he says. “The federal government’s investment in establishing the feasibility of CRISPR cures, I think, ultimately will benefit the taxpayers. Why? Because as we learn more and more about gene editing for rare disease, we will learn more and more how to safely gene edit for common disease.”
Meanwhile, taking lessons from his prior attempt at industrial biotech, Perlstein revived Perlara in late 2021, but this time creating a leaner and stronger company. “It’s decentralized biotech,” he says. “We offer a fundamentally different business model, which is a virtual consultancy.” Perlstein sees Perlara’s role as accelerating the research to find treatments for patients who would otherwise remain under the radar of big pharma. “The whole purpose of the company now is to become the Y Combinator for rare diseases,” Perlstein said. “What Y Combinator did for overlooked founders, I’m doing the same thing with overlooked families.”
In its new format, Perlara no longer has a permanent, physical lab space or full-time employees; it has dispensed with them in favor of geographically distributed pop-up labs and “cure guides” who work as independent contractors with the company. “It’s kind of like Uber,” quips Perlstein, “except that instead of drivers, I have cure guides.” The cure guides get matched with families and form research teams to develop a personalized treatment, much as Chow’s lab took on Jake Carroll’s case.
For now, Perlstein acknowledges, families working with Perlara are largely self-financed. He calls them “the tip of the spear” in the quest for rare disease treatments, which could ultimately become available to other patients. “I’m not trying to make something that only benefits the 1%,” he said. “But I’m not going to ignore the fact that they exist.” Indeed, Maggie’s success with epalrestat led to the launch in December 2022 of a 40-person phase 3 clinical trial. Without the Carmichaels’ efforts, epalrestat’s benefits for CDGs may have remained unknown, even though the genetic and biochemical technology required to find it is accessible in most modern labs.
It remains to be seen whether rare disease drug discovery will be financially sustainable, whether using the Perlara model, which so far relies on funding from families; the n-Lorem model, which draws on corporate donations and advocacy organizations; or the public-funding scenario proposed by Urnov. Other for-profit ventures are springing up to try to accelerate N-of-1 therapies; for instance, Julia Vitarello, Mila’s mother, recently co-founded EveryONE Medicines with investment support from GV and Khosla.
“This is the time for rare diseases,” says Chow. “In the next ten years, I think you’ll see therapies rolling out left and right because it’s just been waiting for the technology to be there.” He points out that rare genetic diseases are far simpler to model in the lab than complex multifactorial conditions like heart disease or cancer. “I think we have all the tools,” he says. “What’s lacking is the funding.”
Read More
Caroline Seydel